Lipid json file format
Contents
Lipid json file format#
To build new hydrogens on a united-atom lipid, we need different informations that are read by buildH in a json file. By default, some standard lipids are present in buildH (in the directory buildh/lipids/). However, it is possible for the user to supply his/her own json file. Here we explain the format of these json files. All images in this page were generated with Pymol.
Generality on the lipid json format#
The convention for naming the json file is Forcefield_Lipid.json. Forcefield obviously specifies the force field and Lipid is the residue name of the lipid in the pdb or gro file. One such name can be for example Berger_POPC.json.
The format of the json file ressembles a Python dictionnary. For example, if we look at the Berger_POPC.json file (already present in buildH), we have the following:
{
"resname": ["POPC", "PLA", "POP"],
"C1": ["CH3", "N4", "C5"],
"C2": ["CH3", "N4", "C5"],
"C3": ["CH3", "N4", "C5"],
"C5": ["CH2", "N4", "C6"],
"C6": ["CH2", "C5", "O7"],
"C12": ["CH2", "O11", "C13"],
"C13": ["CH", "C12", "C32", "O14"],
[...]
"C24": ["CHdoublebond", "C23", "C25"],
"C25": ["CHdoublebond", "C24", "C26"],
"C26": ["CH2", "C25", "C27"],
[...]
"C48": ["CH2", "C47", "C49"],
"C49": ["CH2", "C48", "C50"],
"C50": ["CH3", "C49", "C48"]
}
Each lines has a key: value pattern as in a Python dictionnary. The value ressembles a Python list [value1, value2, ...]. Each couple key: value is separated by a comma. At the beginning and end of the file we have curly braces {}.
Important: note that in the last line (atom "C50"), the comma is not present at the end of the line.
The first line with a key "resname" indicates some possible residue names for the lipid described in the file. Here for example, it can be called "POPC", "PLA" or "POP" in the pdb or gro file. Do not forget the quotes for each element. Thanks to this line, one can then use Berger_POPC, Berger_PLA, or Berger_POP with the -l argument when launching buildH at the Unix command line.
In the the next lines, each key is basically a carbon atom name (between quotes) on which one wants to reconstruct hydrogens. This is the same atom name as found in the pdb or gro file. The corresponding value is a list containing 3 or 4 strings separated by a comma:
The first string can be either
"CH","CH2","CH3"or"CHdoublebond". It indicates to buildH if we want to reconstruct one H, 2 Hs, 3 Hs (sp3 carbon) or one H of a double bond (sp2 carbon) respectively. In fact, it represents the type of carbon on which we want to build new hydrogens.The next strings are called helpers (see below) and are atom names between quotes. We have 2 helpers for
"CH2","CH3"or"CHdoublebond", and 3 helpers for"CH".
So the general syntax is [type of C on which we want to build Hs, name of helper1, name of helper2, ...]. The choice of helpers and their order in the json file depends on the type of carbon. Everything is described below.
CH3#
In the figure below is shown a resconstruction of 3 hydrogens (methyl group) on atom C1. In the json file, it corresponds to the line "C1": ["CH3", "N4", "C5"],. The first helper (helper1) needs to be the one connected to C1 (thus N4), and the second helper (helper2) is connected to N4 and 2 atoms away from C1 (thus C5) along the main chain.
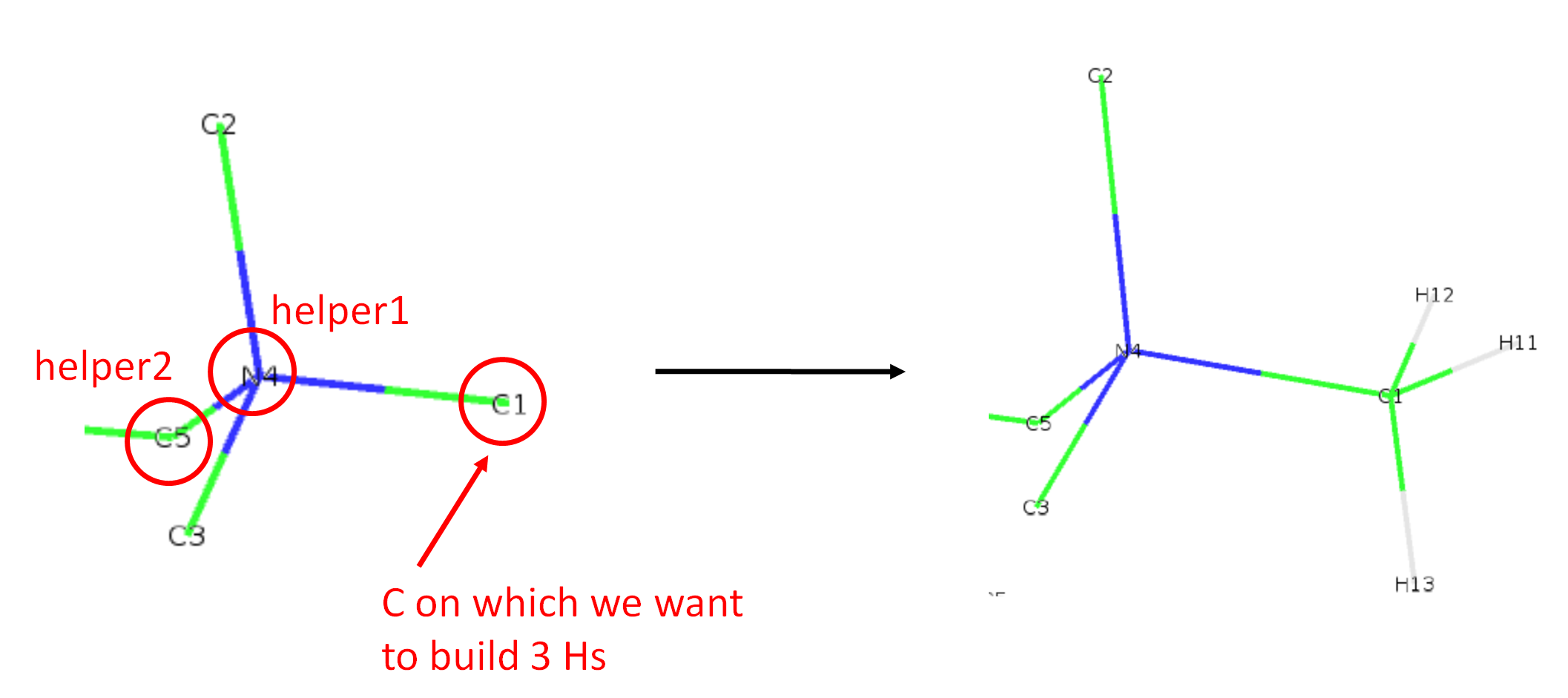
The names of the 3 reconstructed H, here H11, H12 and H13, are infered from the def file supplied with option -d. In this example, Berger_POPC.def was used, the names for the 3 H of C1 were read from the 4th column of these lines:
gamma1_1 POPC C1 H11
gamma1_2 POPC C1 H12
gamma1_3 POPC C1 H13
In this example, the use of C2 or C3 as helper2 would have worked too. However, we decided to use C5 because it stands along the main chain of the lipid.
CH2#
In the figure below is shown the resconstruction of 2 hydrogens (methylene group) on atom C26. On the left is shown a CH2 reconstruction coming from the line "C26": ["CH2", "C25", "C27"], in the json file. "CH2" means we want to reconstruct 2 hydrogens, "C25" is helper1, "C27" is helper2. With C25 being up and C27 being down, the new hydrogens reconstructed are arranged in space so that H261 comes towards us and H262 goes backwards.
On the right, we show the other case where we swapped the order of helper1 and helper2. One can see that the two reconstructed hydrogens are also swapped. This shows that the order of helpers matters for H reconstruction on CH2!
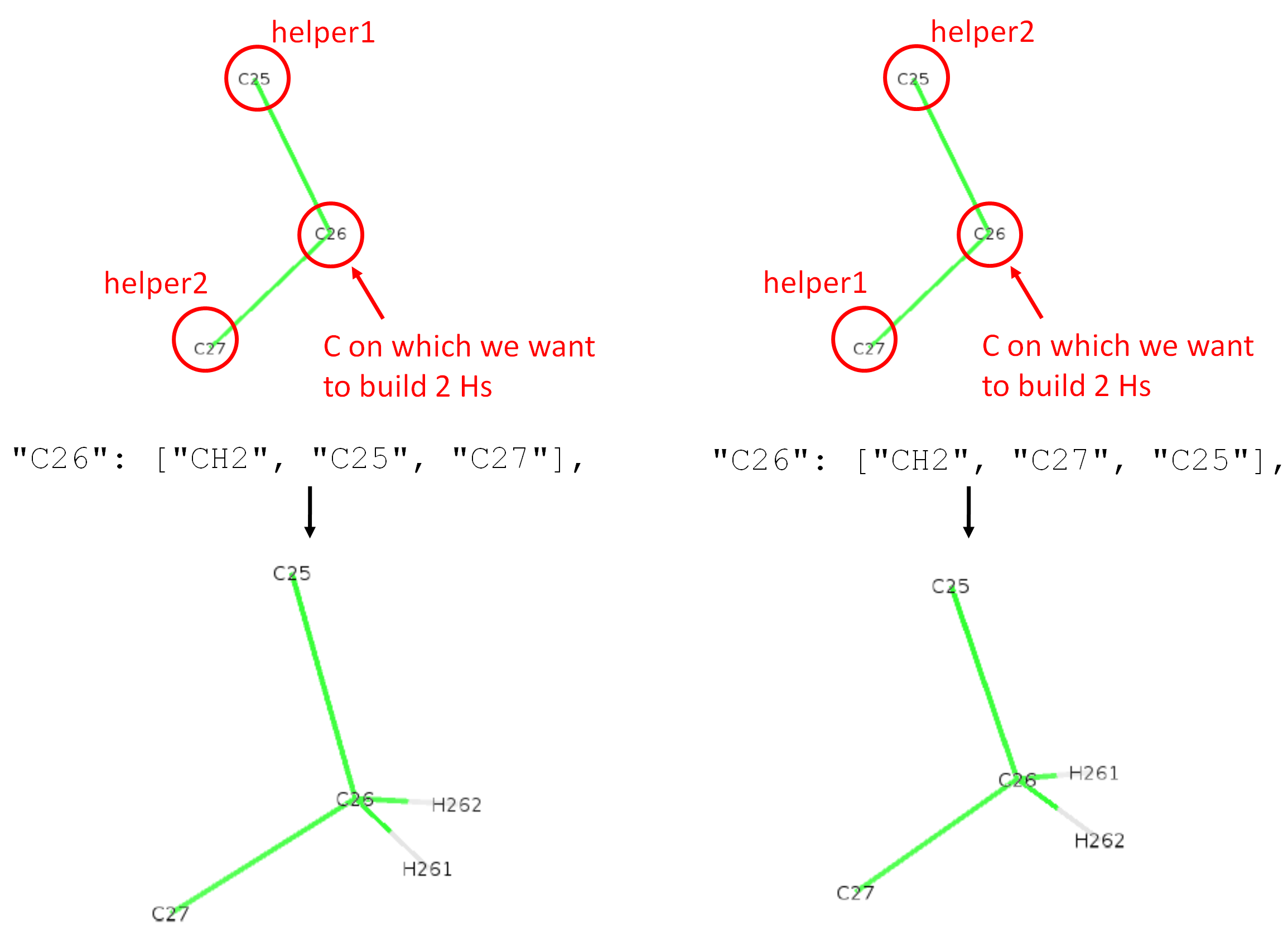
The names H261 or H262 come again from the 4th column of the def file (Berger_POPC.def). The relevant lines are:
oleoyl_C11a POPC C26 H261
oleoyl_C11b POPC C26 H262
TODO: tell which H is pro-R and pro-S.
CH#
For a CH, we want to reconstruct a single hydrogen on a carbon connected to 3 other heavy atoms. In this case, the carbon can be asymetric. This is the case, for example, in phospholipids for the second carbon of the glycerol as shown in the figure below. There is shown the resconstruction of a unique hydrogen on atom C13. In this case, we have 3 helpers which are merely the 3 heavy atoms (C12, C32, and O14) connected to that carbon. Note that the order of helpers in the json file "C13": ["CH", "C12", "C32", "O14"], does not matter in this case. "C12", "C32" and "O14" can be put in any order in this list, the H reconstruction will be strictly identical.
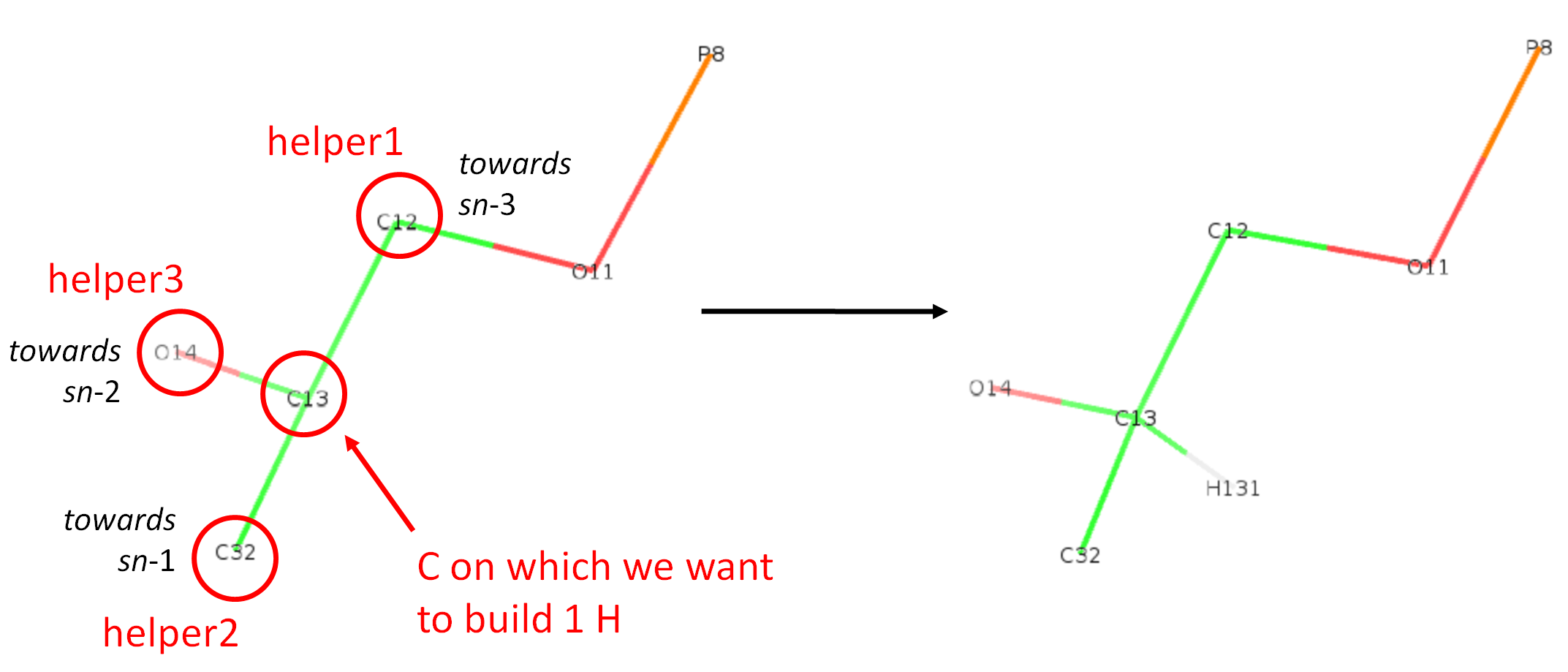
The name H131 comes again from the 4th column of the def file (Berger_POPC.def). The relevant line is:
g2_1 POPC C13 H131
CH of a double bond#
When a carbon is involved in a double bond, we want to reconstruct a single H which respects the sp2 geometry. Below is shown an example on which the double bond stands between C24 and C25 and we want to build the single H on C25. The line in the json file for such a case is "C25": ["CHdoublebond", "C24", "C26"],, where the first string in the list is now "CHdoublebond". The two helpers are C24 and C26 which are the atoms directly bonded to C25. Note that the order of helpers in the list does not matter in this case, "C24", "C26" or "C26", "C24" will work the same.
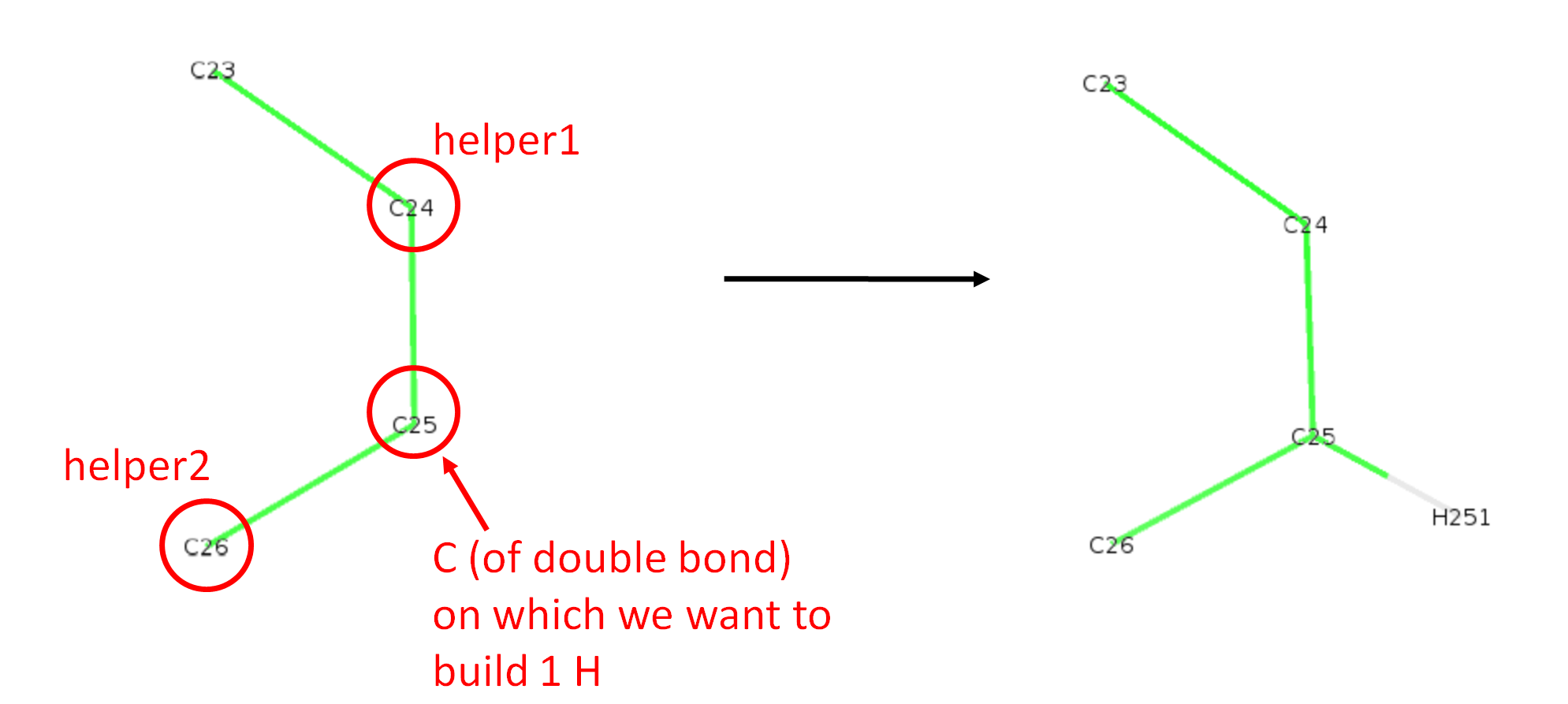
The name H251 comes again from the 4th column of the def file (Berger_POPC.def). The relevant line is:
oleoyl_C10a POPC C25 H251
Summary#
We have explained here the format of the json file which tells buildH what are the carbons on which we want to reconstruct hydrogens and the corresponding helpers. Finally, we draw again your attention about the order of helpers within the json file:
the order of helpers in each list does not matter in the case of a CH or CHdoublebond;
the order of helpers in each list does matter in the case of a CH3 or CH2 reconstruction:
for CH3, helper1 is bonded to the carbon on which we reconstruct hydrogens and helper2 is two atoms away;
for CH2, both helper1 and helper2 are bonded to the carbon on which we reconstruct hydrogens, but their order will determine which reconstructed H is pro-R or pro-S.
A guided example for writing a lipid json file#
We show here how to build your own json file on the simple molecule of butane. We start with a pdb file of the molecule butane.pdb:
ATOM 1 C1 BUTA 1 -1.890 0.170 0.100 1.00 0.00
ATOM 2 C2 BUTA 1 -0.560 -0.550 -0.100 1.00 0.00
ATOM 3 C3 BUTA 1 0.540 0.520 -0.110 1.00 0.00
ATOM 4 C4 BUTA 1 1.910 -0.140 0.100 1.00 0.00
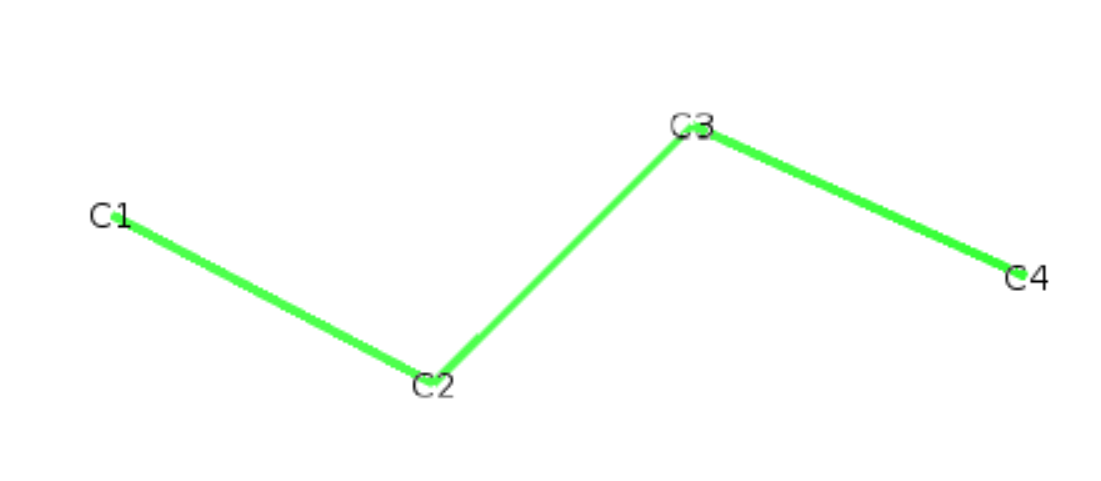
Now we need to build the json file. According to the rules above, we have the following:
C1is of type CH3. Helper1 is connected toC1(thusC2), helper2 is two atoms away (thusC3).C2is of type CH2. Helper1 is the carbon before in the chain (thusC1), helper2 is the atom after in the chain (thusC3).C3is also of type CH2, so following the same rule, helper1 isC2and helper2 isC4.C4is also of type CH3, so following the same rule, helper1 isC3and helper2 isC2.
In summary, this would give the following file:
{
"resname": ["BUTA", "BUT"],
"C1": ["CH3", "C2", "C3"],
"C2": ["CH2", "C1", "C3"],
"C3": ["CH2", "C2", "C4"],
"C4": ["CH3", "C3", "C2"]
}
We have to name it with the convention Forcefield_Residue.json. So let us imagine we use Berger force field, we can call it Berger_BUTA.json.
We also have to create the def file (see here on how to do that). We can use the following Berger_BUTA.def:
butane_C1a BUTA C1 H11
butane_C1b BUTA C1 H12
butane_C1c BUTA C1 H13
butane_C2a BUTA C2 H21
butane_C2b BUTA C2 H22
butane_C3a BUTA C3 H31
butane_C3b BUTA C3 H32
butane_C4a BUTA C4 H41
butane_C4b BUTA C4 H42
butane_C4c BUTA C4 H43
With those 3 files, we can launch buildH:
buildH -c butane.pdb -l Berger_BUTA -lt Berger_BUTA.json -d Berger_BUTA.def -opx butane_wH
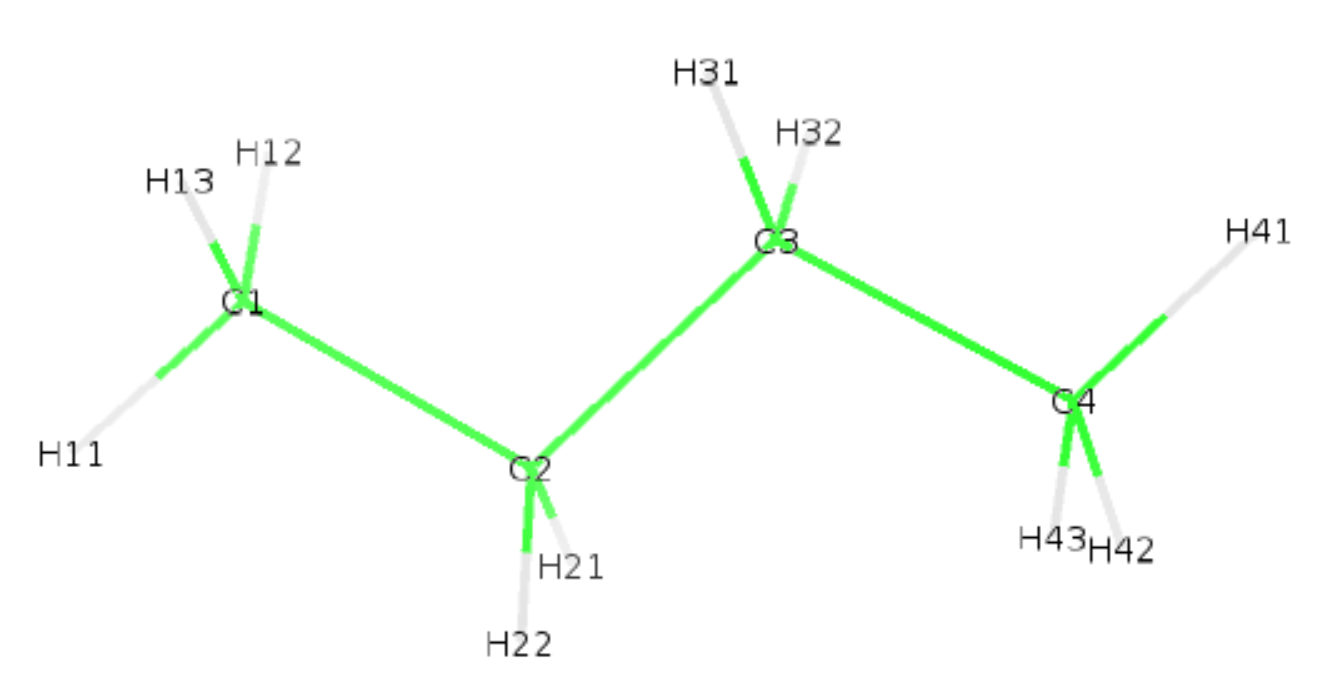
So we used here the option -lt to supply our own Berger_BUTA.json file. We also requested an ouput with option -opx which will generate the pdb with hydrogens butane_wH.pdb. Below is shown the generated pdb and structure.
ATOM 1 C1 BUTA 1 -1.890 0.170 0.100 1.00 0.00 C
ATOM 2 H11 BUTA 1 -2.700 -0.560 0.113 1.00 0.00 H
ATOM 3 H12 BUTA 1 -2.048 0.874 -0.717 1.00 0.00 H
ATOM 4 H13 BUTA 1 -1.872 0.710 1.047 1.00 0.00 H
ATOM 5 C2 BUTA 1 -0.560 -0.550 -0.100 1.00 0.00 C
ATOM 6 H21 BUTA 1 -0.566 -1.088 -1.048 1.00 0.00 H
ATOM 7 H22 BUTA 1 -0.390 -1.253 0.716 1.00 0.00 H
ATOM 8 C3 BUTA 1 0.540 0.520 -0.110 1.00 0.00 C
ATOM 9 H31 BUTA 1 0.356 1.235 0.692 1.00 0.00 H
ATOM 10 H32 BUTA 1 0.531 1.039 -1.069 1.00 0.00 H
ATOM 11 C4 BUTA 1 1.910 -0.140 0.100 1.00 0.00 C
ATOM 12 H41 BUTA 1 2.687 0.625 0.092 1.00 0.00 H
ATOM 13 H42 BUTA 1 2.096 -0.855 -0.702 1.00 0.00 H
ATOM 14 H43 BUTA 1 1.920 -0.658 1.059 1.00 0.00 H
The pdb name of the newly built hydrogens (H11, H12, etc.) were infered from the 4th column of the def file.
Last advices
We showed you a simple example on butane. Although this molecule is very simple, you can see that it is easy to make a mistake. So we recommend to triple check the json file before using it for production. buildH makes for you a lot of checks and will throw an error if something is wrong, but it cannot detect all types of mistakes. Any spelling error on atom names, inversion, etc., may lead to aberrant results. So before going to production, do test on a single molecule and check thoroughly the molecule has all the hydrogens in good place.
The main lipids are already included in buildH (in the directory
buildh/lipids) so you might not need to build your own json file. You can have a list of the supported lipids by invoking buildH with option-h:
$ buildH -h
usage: buildH [-h] -c COORD [-t TRAJ] -l LIPID [-lt LIPID_TOPOLOGY [LIPID_TOPOLOGY ...]] -d DEFOP
[...]
The list of supported lipids (-l option) are: Berger_CHOL, Berger_DOPC, Berger_DPPC, Berger_PLA, Berger_POPC, Berger_POP, Berger_POPE, Berger_POPS, CHARMM36UA_DPPC, CHARMM36UA_DPUC, CHARMM36_POPC, GROMOS53A6L_DPPC, GROMOSCKP_POPC, GROMOSCKP_POPS. More documentation can be found at https://buildh.readthedocs.io.
Last, one other project developed by us, called autoLipMap, can build automatically def and json files for the main known lipids.
In case of problem, you can post an issue on github.