Lipid def file format
Contents
Lipid def file format#
Role of def files#
The def file has three main functions:
Tell buildH which C-H bonds are considered for H reconstruction and order parameter calculation.
Give a generic name to each C-H for the output of the order parameter (the default name for that file is
OP_buildH.out); a generic name can be for examplebeta1orbeta2.Give a specific pdb name to each newly built hydrogen when a trajectory is requested.
Initially, this type of file has been created in the NMRlipids project. Starting from all-atom trajectories, the script calcOrderParameters.py takes such def files as input to infer what C-H are considered for the calculation. But it is also useful for assigning a unique generic name for each C-H along the lipid regardless of the force field considered. For example, the two C-H of of the beta carbon of the choline are called beta1 and beta2 in all PC lipids whatever the force field. Last, it is also useful to specify a specific pdb name to each reconstructed hydrogen.
Many of these .def files can be found on the MATCH repository for all-atom force fields. As in buildH we work with united-atom trajectories, there is a directory def_files where you will find all the def files for the lipids supported.
Format of def files#
The general naming convention of def files on NMRLipids is order_parameters_definitions_MODEL_X_Y.def, where X is the force field and Y the lipid considered. You can name it whatever you like, but we recommend to indicate at least the force field and the lipid. In buildH, the def files present in the repo have mere names folllowing the convention Forcefield_Lipid.def, for example Berger_POPC.def.
Each line represents a given C-H with 4 columns:
Column 1 is the generic name of the order parameter considered. We recommend to use the same convention as those already present in the def_files provided in buildH.
Column 2 is the residue name in the pdb or gro file.
Column 3 is the carbon name in the pdb or gro file for that C-H.
Column 4 is the H name for that C-H, it is used if an output pdb file is requested.
Here is an example for the polar head of Berger POPC:
beta1 POPC C5 H51
beta2 POPC C5 H52
alpha1 POPC C6 H61
alpha2 POPC C6 H62
g3_1 POPC C12 H121
g3_2 POPC C12 H122
g2_1 POPC C13 H131
g1_1 POPC C32 H321
g1_2 POPC C32 H322
Each column has to be separated by any combination of white space(s) (at least one).
Trajectory output and def files#
If an output trajectory (option -opx) is requested, the .def file provided must contain all possible C-H in that lipid (since the whole trajectory with Hs will be reconstructed). The newly built hydrogens will follow the names written in the 4th column of the def file. This option is slow, we do not recommend it if an output xtc file is not wanted.
If no option -opx is used, buildH uses fast indexing. In this case the .def file can contain any subset of all possible C-H pairs. For example, if one wants to get the order parameters of the polar head only (Berger POPC), the .def will be the following:
beta1 POPC C5 H51
beta2 POPC C5 H52
alpha1 POPC C6 H61
alpha2 POPC C6 H62
g3_1 POPC C12 H121
g3_2 POPC C12 H122
g2_1 POPC C13 H131
g1_1 POPC C32 H321
g1_2 POPC C32 H322
Of course, a lower number of Hs to reconstruct will make buildH run faster.
A guided example for writing a lipid def file#
We show here how to build your own def file on the simple molecule of butane. We start with a pdb file of the molecule butane.pdb:
ATOM 1 C1 BUTA 1 -1.890 0.170 0.100 1.00 0.00
ATOM 2 C2 BUTA 1 -0.560 -0.550 -0.100 1.00 0.00
ATOM 3 C3 BUTA 1 0.540 0.520 -0.110 1.00 0.00
ATOM 4 C4 BUTA 1 1.910 -0.140 0.100 1.00 0.00
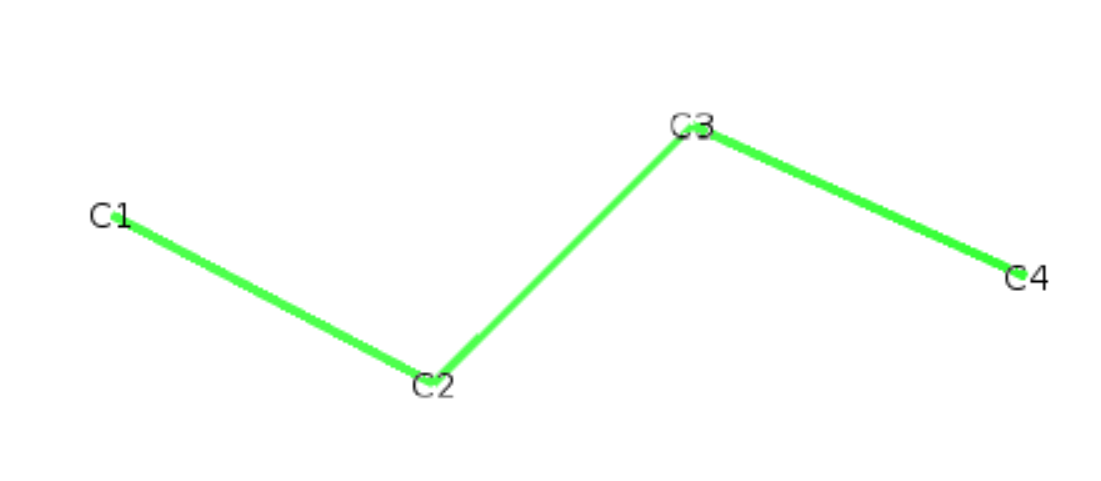
Let us consider first C1 which has 3 hydrogens to reconstruct. We can call the new hydrogens H11, H12 and H13. The residue name is BUTA. Since the butane molecule does not exist in known def files, we can create any generic name we want for each C-H. Following the same model as Berger_POPC.def, we could name them butane_C1a, butane_C1a and butane_C1c. Applying this to all possible C-H, we obtain:
butane_C1a BUTA C1 H11
butane_C1b BUTA C1 H12
butane_C1c BUTA C1 H13
butane_C2a BUTA C2 H21
butane_C2b BUTA C2 H22
butane_C3a BUTA C3 H31
butane_C3b BUTA C3 H32
butane_C4a BUTA C4 H41
butane_C4b BUTA C4 H42
butane_C4c BUTA C4 H43
Following the file naming convention Forcefield_Lipid.def, this file should be called Berger_BUTA.def.
We also have to create the json file (see here on how to do that). We can use the following Berger_BUTA.json:
{
"resname": ["BUTA", "BUT"],
"C1": ["CH3", "C2", "C3"],
"C2": ["CH2", "C1", "C3"],
"C3": ["CH2", "C2", "C4"],
"C4": ["CH3", "C3", "C2"]
}
With those 3 files, we can launch buildH:
buildH -c butane.pdb -l Berger_BUTA -lt Berger_BUTA.json -d Berger_BUTA.def -opx butane_wH
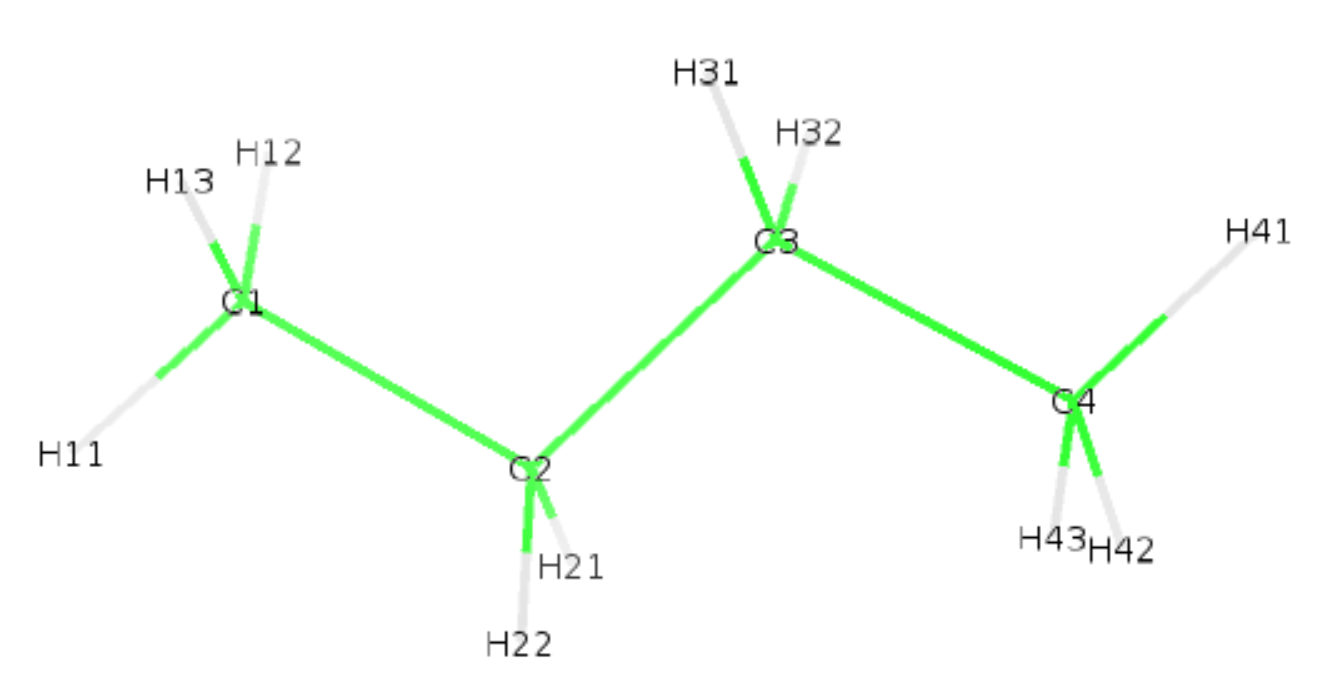
We supplied our newly created def file to buildH with the option -d. We also used our own Berger_BUTA.json file with the option -lt. We requested an ouput with the option -opx which will generate the pdb with hydrogens butane_wH.pdb. Below is shown the generated pdb and structure.
ATOM 1 C1 BUTA 1 -1.890 0.170 0.100 1.00 0.00 C
ATOM 2 H11 BUTA 1 -2.700 -0.560 0.113 1.00 0.00 H
ATOM 3 H12 BUTA 1 -2.048 0.874 -0.717 1.00 0.00 H
ATOM 4 H13 BUTA 1 -1.872 0.710 1.047 1.00 0.00 H
ATOM 5 C2 BUTA 1 -0.560 -0.550 -0.100 1.00 0.00 C
ATOM 6 H21 BUTA 1 -0.566 -1.088 -1.048 1.00 0.00 H
ATOM 7 H22 BUTA 1 -0.390 -1.253 0.716 1.00 0.00 H
ATOM 8 C3 BUTA 1 0.540 0.520 -0.110 1.00 0.00 C
ATOM 9 H31 BUTA 1 0.356 1.235 0.692 1.00 0.00 H
ATOM 10 H32 BUTA 1 0.531 1.039 -1.069 1.00 0.00 H
ATOM 11 C4 BUTA 1 1.910 -0.140 0.100 1.00 0.00 C
ATOM 12 H41 BUTA 1 2.687 0.625 0.092 1.00 0.00 H
ATOM 13 H42 BUTA 1 2.096 -0.855 -0.702 1.00 0.00 H
ATOM 14 H43 BUTA 1 1.920 -0.658 1.059 1.00 0.00 H
Last advices
We showed you a simple example on butane. Although this molecule is very simple, you can see that it is easy to make a mistake. Although this def file is less critical than the json lipid file, we recommend to double check it before using it for production. buildH makes for you a lot of checks and will throw an error if something is wrong, but it cannot detect all types of mistakes. Any spelling error on atom names, inversion, etc., may lead to nonsense results. So before going to production, do test on a single molecule and check thoroughly on a couple of examples whether the output looks right.
The main lipids are already included in buildH (in the directory
buildh/lipids) so you might not need to build your own def file. You can have a list of the supported lipids by invoking buildH with option-h:
$ buildH -h
usage: buildH [-h] -c COORD [-t TRAJ] -l LIPID [-lt LIPID_TOPOLOGY [LIPID_TOPOLOGY ...]] -d DEFOP
[...]
The list of supported lipids (-l option) are: Berger_POP, Berger_POPC, Berger_PLA, Berger_POPE, CHARMM36_POPC.
[...]
Last, one other project developed by us, called autoLipMap, can build automatically def and json files for the main known lipids.
In case of problem, you can post an issue on github.